
АВТОР: Джилл Рамзби (Gill Rumsby)
Отдел клинической биохимии, Городская лондонская больница, улица Уитфилд 60, Лондон W1T 4EU, Великобритания (Clinical Biochemistry, UCL Hospitals, 60 Whitfield St, London W1T 4EU, United Kingdom)
ОСНОВНЫЕ ПОЛОЖЕНИЯ:
- Описана молекулярная основа 4 унаследованных почечнокаменных болезней.
- Причина образования камней зависит от типа почечнокаменной болезни.
- Важность выявления таких нарушений имеет очень большое значение для здоровья почек и здоровья других родственников.
КЛЮЧЕВЫЕ СЛОВА: генетика, камни, гипероксалурия, цистинурия, дистальный почечноканальцевый ацидоз, дефицит аденин-фосфорибозилтрансферазы.
АННОТАЦИЯ
Ренальные камни – весьма распространенная проблема. Они, как правило, являются вторичными, по отношению к факторам риска, которые влияют на растворимость веществ в мочевыводящих путях. Первичные, то есть генетические, причины не столь распространены, но, тем не менее, о них нужно знать, чтобы прописывать соответствующее лечение и учитывать риски, угрожающие другим членам семьи.
В данной статье представлен краткий обзор исследования ренальных камней с биохимической точки зрения, он сосредоточен на проблемах, которые могут возникнуть в дальнейшем лечении.
Генетические причины, вызывающие появление ренальных камней, связаны с: (I) нарушением метаболического пути, (II) наличием нерастворимых продуктов, (III), нарушением транспортировки пищи (IV) и почечноканальцевым ацидозом. Все эти проблемы описаны по отношению к нарушениям работы и дефициту аденин фосфорибозилтрансферазы (АФРТ), дефициту первичной гипероксалурии, цистинурии и аутосомно-доминантного дистального почечноканальцевого ацидоза.
Камни в почках (нефролитиаз) – весьма распространенная проблема. Согласно различным оценкам, этой болезнью страдает от 6 до 15% населения в странах западного мира [1]. В большинстве случаев она вызвана факторами окружающей среды: родом деятельности человека, питанием, низким потреблением жидкости, отсутствием каменных ингибиторов, введением нерастворимых препаратов, например антиретровирусных средств, или же связаны с приобретенными болезнями, такими как гиперпаратиреоз, либо анатомическими факторами, например, подковообразным строением почки. Нефрокальциноз – расстройство, связанное с нарушением работы почек. Оно может указывать на наличие кальция в почках. В небольшой, но значимой части, нефрокальциноз и почечнокаменная болезнь являются результатом наследственных заболеваний, аутосомно-доминантных, рецессивных нарушений, или нарушений, связанных с Х-хромосомой. Выявление таких болезней имеет крайне важное значение для лечения и определения рисков для других членов семьи. В этом обзоре основное внимание уделяется наследственным заболеваниям почечнокаменной болезнью. Обзор не охватывает всех вопросов – это невозможно, поскольку список отклонений постоянно растет. В работе достаточно хорошо освещены причины метаболических расстройств, вызванные: (I) нарушением метаболического пути, (II) наличием нерастворимых продуктов, (III) нарушением транспортировки пищи и (IV) почечно-тубулярным ацидозом.
Подозрение на наследственную причину возникновения почечнокаменной болезни может возникнуть под влиянием одного из следующих факторов: проблема возникла в молодом возрасте, камни являются рецидивирующими, это двусторонние камни, семейная история подтверждает вероятность наличия наследственной проблемы. Начальные исследования можно выполнить (Таблица 1) в большинстве клинических биохимических лабораторий либо на дому (большинство тестов), либо путем направления в специализированные центры (в случае таких заболеваний как оксалат, цитрат, цистеин, первичная гипероксалурия метаболитов). Анализ камней в почках, если таковые имеются, весьма полезен [2], но полученные нами данные свидетельствуют о том, что не все лаборатории гарантируют надежный результат и в некоторых случаях неправильно определяют тип каменной болезни, не могут определить тип редкого камня и не предоставляют информации о составе и различных компонентах. Тесты, которые используют физический анализ, например, инфракрасный анализ, с большей вероятностью дадут надежные результаты. Рентгенографический анализ поможет определить, содержат ли камни кальций (рентгеноконтрастный), или являются рентгенопроницаемыми. Последнее указывает на наличие мочевой кислоты или другого пуринового материала, или материала со слабой радио-плотностью, как видно по цистиносодержащим камням. Почечнокаменная болезнь детей исследуется с помощью метаболического профиля, но, к сожалению, такое исследование не применяется в работе со взрослыми – часто камни лечат чисто физически, без сопровождающих биохимических исследований. Даже для детей диагностика занимает довольно много времени, может сопровождаться задержками, которые в долгосрочной перспективе вызывают травмирующее действие на почку. Существует старая поговорка, которая гласит: "Не бывает бывшего камня". Рецидивирующие камни – типичное явление для пациентов с почечнокаменной болезнью, и, к сожалению, проблема не изучается более подробно.
Камни в почках могут быть прямым следствием потери фермента, что приводит к накоплению нерастворимого вещества, в других случаях дефект гена предрасполагает индивида к почечным камням, как, например, в случае с почечноканальцевым ацидозом. Общее количество вовлеченных в этот процесс генов пока неизвестно, но недавно было проведено исследование 30 известных генов, которые влияют на формирование камней у пациентов, проходящих лечение в клинике, специализирующейся на ренальных камнях (за исключением первичного системного заболевания и камней, появление которых связано с употреблением наркотиков), в результате выявлены мутации в 15% изученных случаях (21% у детей и 11% у взрослых)[3]. Цистинурия, вызванная мутациями в SLC7A9 оказалась самым распространенным заболеванием.
Таблица 1
Исследование почечнокаменной болезни.
| Первичное
исследование Вторичные исследования | моча
за 24 часа (обычная) Объем креатинин урат протеин
уровень
pH Цистин
(капельный тест)
Цистин (количественное определение) если капельный
тест оказался положительным | моча
за 24 часа (сбор кислоты) Объем Креатинин Кальций Магнезий Цитрат Оксалат Первичная
гмпероксалурия метаболитов (гиколяты, глицераты, HOG и DHG) |
НАРУШЕНИЕ МЕТАБОЛИЧЕСКОГО ПУТИ
Нарушения метаболизма пуринов, приводящие к дефициту ферментов аденин-фосфорибозилтрансферазы (Aprt) и ксантин-дегидрогеназы (КДГ) иллюстрирует этот тип нарушения обмена веществ. В обоих случаях нарушение приводит к накоплению веществ, предшествующих блокированию ферментов. В случае дефицита аденин-фосфорибозилтрансферазы, аденин накапливается и перетравливается КДГ до 2,8-дигидроксиаденина (ДГА); поскольку отсутствие КДГ приводит к накоплению ксантина. ДГА и ксантин – нерастворимые вещества, которые образуют почти чистые, рентгенопрозрачные камни. Неспецифический влажный химический метод, который использует сокращение реагента Фолина (фосфотунгстат/фосфоромолибдат) для обнаружения мочевой кислоты также дает положительный результат под влиянием ДГА и ксантина и поэтому анализ почечных камней может дать некорректный результат, если не использовать физический метод (Рис. 1). Более надежный результат можно получить с помощью спектрометрии (Рис. 1), но даже в этом случае потребуется опытный оператор, который сможет подтвердить тип камня. КДГ может вызывать очень низкий уровень сыворотки в мочевой кислоте, что, скорее всего, связано с тем, что пациент не получает ингибиторы уриказы. Оба расстройства могут быть подтверждены путем измерения соответствующих пуриновых метаболитов в моче и для аденин-фосфорибозилтрансферазы, измерение ферментативной активности в красных клетках [4].
Оба заболевания являются аутосомно-рецессивными. Фермент аденин-фосфорибозилтрансфераза кодируется АФРТ, геном на длинном плече хромосомы 16 (16q24) [5]. До настоящего времени описаны более 40 мутаций с некоторыми общими характеристиками для конкретных групп населения, например, p.Met136Thr с менее чем 10% от нормальной активности [6] обнаружен у более чем 79% пациентов из Японии [7]. Наиболее распространенной мутацией (40%) среди французов является c.400þ2dup, что приводит к неправильному сращиванию [8], тогда как c.194A> T (p.Asp65Val) является наиболее распространенным среди кавказцев и особенно часто встречается в Исландии [9]. Гетерозиготность расстройства, по оценкам ферментных исследований, составляет 1/100 [10] указывая на то, что дефект не может быть настолько редким, как считалось ранее. Это предположение подтверждается выводом о мутации c.400þ2dup у 1% здоровых новорожденных [8], что может проявиться позднее, либо камень могут неправильно диагностировать в результате неточного анализа, как камень из мочевой кислоты. Конечной стадией такого расстройства является почечная недостаточность, которую не устранить даже трансплантацией почки.
НАРУШЕНИЕ ОБМЕНА НЕРАСТВОРИМЫХ ПРОДУКТОВ
Первичная гипероксалурия (ПГ) является прекрасной иллюстрацией камней данного типа. Существуют три известных типа ПГ, каждый из которых приводит к метаболизму предшественников в нерастворимом оксалате кальция, который выпадает в осадок в почках в виде нефрокальцинозы или почечных камней. Камни, как правило на 100% состоят из оксалата кальция, часто моногидрата, что указывает на быстрое образование, и в случаях ПГ1 имеет необычную морфологию [11], также он может смешиваться с фосфатом кальция. Все три типа, ПГ1, ПГ2 и ПГ3 - это аутосомно-рецессивные расстройства, которые вызваны дефицитом аланина: ферменты глиоксилатной аминотрансферазы, глиоксилат/оксипировиноград редуктазы и гидроксиоксоглютарат альдолазы (Рис.2), кодируемые генами AGXT, GRHPR и HOGA1 соответственно. Экскреции оксалатов мочи похожи при всех трех расстройствах, но могут быть различными [12]. В качестве приемлемого уровня предложена концентрация оксалата мочи свыше 0,7 ммоль/день [13]. При таком уровне следует рассматривать основную причину (у детей результат должен быть выражен/1,73 м2), но это не обязательно исключает вторичные причины, например бариатрическую хирургию [14], или хронический панкреатит, более низкая концентрация не исключает ПГ, поэтому если все еще есть подозрение, необходимо провести дополнительные тесты. Выявление 4-гидрокси-2-оксоглутарата (HOG) и дигидроксиглютората в моче у пациентов с ПГ3 [15, 16] привело к диагностированию первичной гипероксалурии метаболитов (OCM) – это следующий шаг в исследовании заболевания (Таблица 1). Этот анализ может обеспечить дополнительную поддержку для различных первичных гипероксалурий: повышенный гликолят (найденный в 70% ПГ1), глицерат (найден в более 95% случаев ПГ2) и дигидроксиглюторат (во всех случаях ПГ3 до настоящего времени), поэтому необходимо сосредоточиться на генетическом тестировании. HOG является менее надежным маркером, так как он неустойчив, если не собирается в подкисленной моче и поэтому может дать ложный отрицательный результат [17]. ПГ1 является наиболее распространенным из всех трех заболеваний (около 80% случаев по нашему опыту [18]), на ПГ2 и ПГ3 приходится около 10% случаев. Возраст возникновения данных образований – это, как правило, раннее детство, примерно 5 лет [19].
AGXT происходит в двух аллельных формах, большой и малой аллелях, последняя кодирует белок с аминокислотной заменой, p.Pro11Leu, который имеет приблизительно 60% основной активности [20] и представляет собой слабую митохондриальную последовательность [21,22]. Малая аллель случается с частотой примерно 20% у европейцев и гораздо реже у афро-американцев [23]. Мутации, происходящие на фоне малой аллели, как правило, имеют более низкую активность и стабильность [24,25], по крайней мере, в лабораторных условиях. Более 170 мутаций, описанных (см www.uclh.nhs.uk/phmd~~dobj) в большинстве случаев, приводят к значительному уменьшению или полной потери ферментативной активности [20]. Большинство из них – одиночные нуклеотидные изменения с c.508G> A (p.Gly170Arg), происходящие в 30% случаях мутантных аллелей среди всех пациентов [20]. Этот мутант сохраняет значительную активность в пробирке, но димеризация белка замедляется, в результате чего фермент не попадает в митохондрии и не может выполнять свою роль в глиоксилатной детоксикации [26]. У пациентов с этой мутацией и вариантом p.Phe152Ile бывает реакция на пиридоксин [27] (пиридоксин является кофактором для аланина: глиоксилатная аминотрансфераза). В некоторых случаях, в сочетании с гидратацией, пиридоксин приводит к экскреции оксалатов мочи в пределах нормы [27] и может оказать влияние на исход трансплантации [28]. Биологическая вариация оксалата мочи высока и, следовательно, требует уменьшения, по крайней мере, на одну треть, чтобы представлять значительные изменения [29].
Мутации в GRHPR весьма разнообразны, но есть два общих изменения и оба они ведут к отсутствию сращивания. Первое c.103delG [30,31] характерно для кавказцев, составляет 37% мутаций в нашей группе пациентов [31] и имеет несущую частоту 1:375 [23]. Второе представляет собой 4 базовые удаления пар с [403_404þ2delAAGT] [30,31] и характерно преимущественно для азиатов. Оно вызывает отсутствие расщепления у mRNA и составляет 18% мутантных аллелей [31]. ПГ3, результат мутации inHOGA1 было недавно описано в [15], но есть две общие патологические вариации. Одна из них, удаление a 3bp (c.944_946delAGG, p.Glu315del) [15] характерна для еврейского населения ашкенази. Вторая, c.700þ5G>T обнаружена в нескольких группах пациентов [15,19] и может вызывать отсутствие сращивания в РНК печени [19] и в пробирке [15]. Из анализа общедоступных баз данных касательно экзом, частота этой мутации (1:165) гораздо выше, чем можно было бы ожидать для частоты этого заболевания [23]. В нашей группе пациентов мы обнаружили 3 гетерозиготы по данной мутации у пациентов с подтвержденным ПГ1 [32]. Этот вывод имеет последствия для генетической диагностики (см ниже).
Генетическое тестирование доступно для всех трех заболеваний. Оно подтверждает биохимическое тестирование, позволяет провести пренатальную диагностику и подтвердить/исключить заболевания у других членов семьи. Этот последний пункт имеет большое значение, так как все три типа болезней могут быть клинически скрытыми до взросления, хотя такие пациенты подвержены риску внезапного возникновения почечной недостаточности. Трансплантация почки является частью плана по лечению ПГ1 и ПГ2. В первом случае, как правило, после трансплантации печени [33], рекомендуется использование родственной живой почки одного из членов семьи - она нуждается в тщательном исследовании у потенциальных доноров органов. Генетическое тестирование с учетом выводов касательно следующего поколения, возникновение одной мутации и гипероксалурия больше не могут рассматриваться как окончательная диагностика для первичной гипероксалурии. Другая причина наследования камней с оксалатом кальция недавно была описана в результате мутаций в SAT1, переносчика сульфатоксалата, кодируемого SLC26A1 [34]. Гипероксалурия присутствует, но не является чрезмерной. Тем не менее, это открытие дает объяснение для пациентов с подозрением на унаследованную гипероксалурию, в случае если три известные формы ПГ уже исключены.
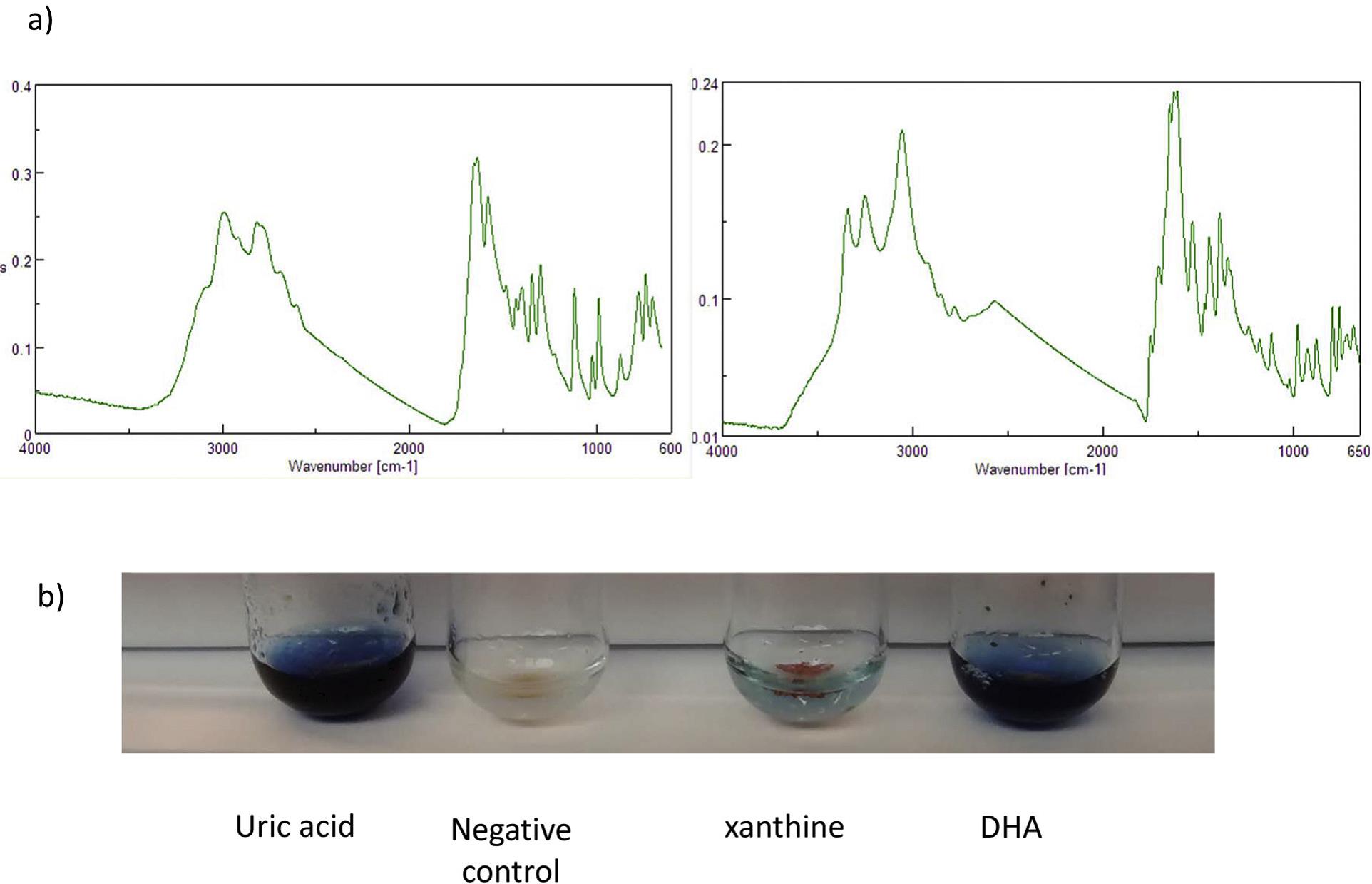
Рисунок 1. а) преобразование Фурье в инфракрасном спектре мочевой кислоты (слева) и ДГК (справа) показывает очевидную разницу спектральных свойств. б) реагент Фолина (в фосфотунгстате/фосфоромолибдате) в тесте для мочевой кислоты также показывает положительные результаты (синяя окраска) с ксантином и ДГК.
ПРОБЛЕМА ТРАНСПОРТИРОВКИ
Аминокислота цистин, наряду с другими катионными аминокислотами, реабсорбируется, главным образом через апикальную мембрану проксимальных канальцев в почках и тонкокишечных эпителиальных клетках с помощью переносчика гетеромерной аминокислоты (ГАК) в обмен на внутриклеточные нейтральные аминокислоты. ГАК состоит из тяжелой и легкой цепи, соединенных на внеклеточной поверхности, дисульфидной связью. Транспортировщик цистина состоит из тяжелой цепи rBAT, кодируемой SLC3A1 [35] и b0,þAT, легкая цепь кодируется SLC7A9 [36]. Мутации в гене могут привести к состоянию цистинурии и характеризуются высокой экскрецией цистина, лизина, орнитина и аргинина. Фракционная экскреция цистина обычно составляет 0,4%, но эта цифра возрастает до 100% при цистинурии. Есть также значительная потеря других основных аминокислот, но цистин является наименее растворимым и, следовательно, приводит к патологии – на него приходится до 8% у детей [37]. 90% цистина реабсорбируется в секциях S1 и S2 проксимально-извитых канальцах, распределение rBAT и легкой цепи b0,þAT не является равномерным; rBAT является наиболее распространенным в сегменте S3, но реже встречается в сегментах S2 и S1. b0.þAT демонстрирует обратное распределение и в основном содержится в S1 [38]. Другая легкая цепь белка, переносчика аспартата/глутамата 1 (AGT1) была недавно описана. Она действует совместно с rBAT в других частях проксимального канальца и может абсорбировать остаточный цистин в проксимальных канальцах [39]. Мутации в гене, кодирующем этот белок, потенциально могут быть другой причиной цистинурии.
Гены SLC3A1 и SLC7A9 находятся в хромосомах 2 [40] и 19 [41] соответственно. Были описаны мутации в обоих генах, но чаще всего в SLC7A9, согласно оценкам – примерно у 11% взрослых, и в 21% случаев у детей [3]. Есть три фенотипа, а именно тип А, где оба аллеля SLC3A1 мутировали, тип B вызванный мутациями в SLC7A9 и тип AB, где одна мутация находится в одном гене, а другая в другом. Гетерозигота для мутаций в SLC7A9 демонстрирует повышенное выделение цистина. Диагностика заболевания требует первоначального скрининг-теста мочи по цвету с образованием нитропруссида и цианида натрия. Такой скрининг может давать ложные положительные результаты с гомоцистеином и серосодержащими препаратами и, следовательно, за качественным анализом должен следовать количественный анализ аминокислот мочи. Гомозигота для цистинурии выделяет > 1300 ммоль цистина за день (нормальный уровень <100).
Выполнен генетический анализ соответствующих генов среди большого количества пациентов, являющихся представителями разных этнических групп. p.Met467Thrvariant - это наиболее распространенный тип мутации (30% мутаций SLC3A1 аллелей, в то время как p.Gly105Arg отвечает за 20% мутаций в SLC7A9 (для просмотра воспользуйтесь ссылкой [42]). Остается спорным вопрос о том, требуется ли для подтверждения диагноза выполнять генетический анализ во всех случаях, поскольку это не влияет на лечение заболевания, но дает возможность подтвердить/исключить заболевание у других членов семьи. Во время лечения этого заболевания можно использовать ощелачивание мочи с цитратом калия для увеличения растворимости цистина. Такое лечение плохо переносится пациентами. Также используется агрессивная гидратация - она требует того, чтобы пациенты выпивали, по меньшей мере, 3 л воды в течение дня и ночи. Это помогает удерживать концентрацию цистина в моче ниже 1200 ммоль/л. Цистиновые связывающие препараты - весьма доступны, например, пеницилламин и тиопронин. Цистиновые камни весьма устойчивы к экстракорпоральной ударно-волновой литотрипсии и, следовательно, если вызывают закупорку, должны удаляться хирургическим путем. Почечная недостаточность не является редкостью для цистинурии – согласно одному исследованию она составляет 64% случаев хронической почечной недостаточности во второй стадии [43].
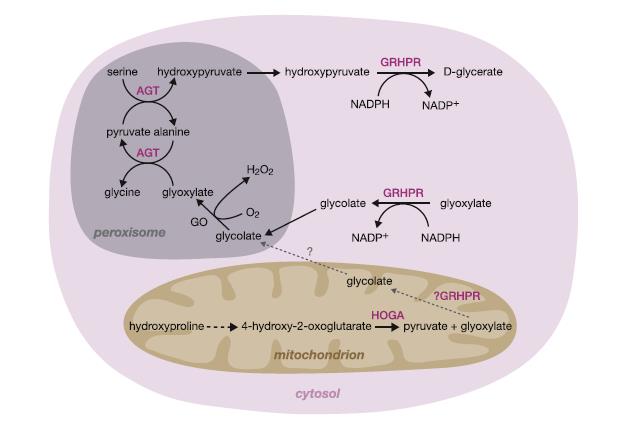
Рисунок. 2. Обмен веществ глиоксилата в гепатоцитах. Глиоксилат метаболизируется аланином: глиоксилат аминотрансферазы (AGT) в пероксисоме и совершается глиоксилатом/гидроксипироватом редуктазы (GRHPR) в цитозоле. Дефицит этих ферментов вызывает ПГ1 и ПГ2 соответственно. В обоих случаях избыток глиоксилатна метаболизируется в оксалат лактатдегидрогеназу. Гидроксиоксоглютарат альдолазы (HOGA) в митохондриях является дефективным ПГ3. Механизм гипероксалурии в этом расстройстве до сих пор до конца неясен, но может отражать распад HOG.
АЦИДОЗ ПОЧЕЧНЫХ КАНАЛЬЦЕВ
Почечные канальцы играют важную роль в выведении воды и ионов Hþ. Большинство нарушений приобретаются, например, из-за избытка ADH в ответ на боль; отложение миеломы приводит к ацидозу почечных канальцев. Однако есть некоторые первичные расстройства в клетках почечных канальцев, которые связаны с образованием камней в почках. В случае ацидоза почечных канальцев (RTA), образование камней происходит по трем причинам. Во-первых внутриклеточный ацидоз приводит к увеличению реабсорбции цитрата в проксимальных канальцах. Цитрат хелатов кальция является естественным ингибитором кристалла и образования камней. Во-вторых, кальций высвобождается из костей, как следствие системного ацидоза. В-третьих, RTA характеризуется наличием щелочи в моче, что способствует осаждению фосфата кальция. Один из примеров доминантно наследуемой дистальной RTA вызван мутациями в базолатеральном анион-обменнике AE1 (Cl/НСО3-), трансмембранном белке, который кодируется SLC4A1 и расположен на хромосоме 17 [44]. Этот ген приводит к двум транскриптам от различных стимуляторов [45]. Более длинный транскрипт, который включает в себя дополнительные 65 аминокислот на терминале N, выражен в мембране эритроцита, в то время как более короткий транскрипт находится в интеркалированных клетках почечных канальцев [46]. Почка AE1 выражена на базолатеральной стороне а-интеркалированных клеток в дистальных почечных канальцах. Фенотип болезни различен: от серьезного образования камней с задержкой роста до скрытого ацидоза и нефрокальциноза. Во любом случае, рН мочи не может окисляться ниже pH 5,5. Гипокалиемия также связана с этим расстройством.
Главное средоточие мутации описано в Arg589 - на эту аминокислоту влияют три мутации (p.Arg589His, p.Arg589Ser, p.Arg589Cys) [47]. Кроме того, кластер делеционных мутаций в пределах от 16 до 61 был недавно описан на примере иранских пациентов и экзонов 11 и 15 в гене [48]. Мутантные белки, выраженные в ооцитах Ксенопсуса, демонстрируют нормальную транспортировку аниона [47], но выражение в поляризованных клетках показало, что эти белки сохранены внутри клеток [49], а в некоторых случаях быстро деградируют [50]. Другие мутанты, в частности p.Arg901X, p.Met909Thr и p.Gly609Arg, выражены в апикальных и базальных мембранах поляризованных клеток [49,51e53]. Мутация p.Arg901X возникает из-за потери последних 11 аминокислот белка [54] и служит доказательством последовательности в карбокситерминальном белке [52], хотя этот сигнал не является достаточным сам по себе, а также может содержать N-терминальные последовательности, необходимые для эффективного нацеливания на базальной мембране [53]. Вопрос заключается в том, как эти мутации вызывают доминантно наследуемые болезни ввиду явного отсутствия влияния на транспортировку аниона. Белок представляет собой димер, и, таким образом, можно было бы ожидать, что он сформирует гетеродимеры одной нормальной и одной мутантной субъединицы с секвестрацией гетеродимера внутриклеточно в случае мутаций p.Arg589His и Gly701Asp [49,50]. Непопадание гетеродимера на апикальную мембрану приводит к выражению в неправильном месте мембраны, бикарбонатой экскреции в просвет, что фактически сводит на нет секрецию кислоты [49].
ВЫВОД
Как уже упоминалось во введении, эта статья не является всеобъемлющей. Она посвящена вопросу того, как молекулярная генетика оживила область изучения почечнокаменной болезни, улучшив диагностику и увеличив наши базовые знания о физиологических процессах. В ближайшие несколько лет, несомненно, будет выявлено больше генетических причин образования камней в почках, так геномные методы активно применяются для изучения причин образования камней и составления прогнозов для членов семьи. До сих пор не изучено, почему болезнь протекает сложнее у отдельных пациентов, хотя степень метаболической аномалии та же. Для того, чтобы ответить на этот вопрос, вероятно, потребуется сочетать методы генетики, протеомики, химии и биохимии.
СПИСОК ЛИТЕРАТУРЫ
[1] G. Bihl, A. Meyers, Recurrent renal stone disease - advances in pathogenesis and clinical management, Lancet 358 (2001) 651e656.
[2] J. Cloutier, L. Villa, O. Traxer, M. Daudon, Kidney stone analysis: 'Give me your stone, I will tell you who you are!, World J. Urol. 33 (2015) 157e169.
[3] J. Halbritter, M. Baum, A.M. Hynes, et al., Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis, J. Am. Soc. Nephrol. 26 (2015) 543e551.
[4] H.A. Simmonds, 2,8-dihydroxyadenine lithiasis, Clin. Chim. Acta 160 (1986) 103e108.
[5] A. Fratini, R.N. Simmers, D.F. Calleln, et al., A new location for the human adenine phosphoribosyltransferase gene (APRT) distal to the haptoglobin (HP) and fra(16)(q23) (FRA16D) loci, Cytogenet Cell Genet. 43 (1986) 10e13.
[6] Y. Hidaka, S.A. Tarie, N. Kamatani, W.N. Kelley, T.D. Palella, Human adenine phosphoribosyltransferase deficiency, J. Clin. Invest. 81 (1988) 945e950.
[7] N. Kamatani, M. Hakodo, S. Otsuka, H. Yoshikawa, S. Kashiwazaki, Only three mutations account for almost all defective alleles causing adenine phosphoribosyltransferase deficiency in Japanese patients, J. Clin. Invest. 90 (1992) 130e135.
[8] G. Bollee, C. Dollinger, L. Boutaud, et al., Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency, J. Am. Soc. Nephrol. 21 (2010) 679e688.
[9] V.O. Edvardsson, R. Palsson, I. Olafsson, G. Hjaltadottir, T. Laxdal, Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland, J. Clin. Invest. 38 (2001) 473e480.
[10] H.A. Simmonds, K.J. Van Acker, Adenine phosphoribosyltransferase defi- ciency: 2,8-dihydroxyadenine lithiasis, in: J.B.W.J.B. Stanbury, D.S. Fredrickson, J.L. Goldstein, M.S. Brown (Eds.), The Metabolic Basis of Inherited Disease, fith ed., McGraw-Hill, 1983, pp. 1144e1156.
[11] M. Daudon, L. Estepa, B. Lacour, P. Jungers, Unusual morphology of calcium oxalate calculi in primary hyperoxaluria, J. Nephrol. 11 (1998) 51e55.
[12] O. Clifford-Mobley, C. Tims, G. Rumsby, The comparability of oxalate excretion and oxalate:creatinine ratio in the investigation of primary hyperoxaluria:a review of data from a referral centre, Ann. Clin. Biochem. 52 (2015) 113e121.
[13] D.S. Milliner, The primary hyperoxalurias: an algorithm for diagnosis, Am. J. Nephrol. 25 (2005) 154e160.
[14] R. Kumar, J.C. Lieske, M.L. Collazo-Clavell, et al., Fat malabsorption and increased intestinal oxalate absorption are common after roux-en-Y gastic bypass surgery, Surgery 149 (2011) 654e661.
[15] R. Belostotsky, E. Seboun, G.H. Idelson, et al., Mutations in DHDPSL are responsible for primary hyperoxaluria type III, Am. J. Hum. Genet. 87 (3) (2010) 392e399.
[16] R. Belostotsky, J. Pitt, Y. Frishberg, Primary hyperoxaluria type III-a model for studying perturbations in glyoxylate metabolism, J. Mol. Med. Berl. 90 (2012) 1497e1504.
[17] O. Clifford-Mobley, L. Hewitt, G. Rumsby, Simultaneous analysis of urinary metabolites for preliminary identification of primary hyperoxaluria, Ann. Clin. Biochem. 53 (2016) 485e494.
[18] P. Cochat, G. Rumsby, Primary hyperoxaluria, N. Engl. J. Med. 369 (2013) 649e658.
[19] E.L. Williams, D. Bockenhauer, W.G. Van't Hoff, et al., The enzyme 4-hydroxy- 2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3, Nephrol. Dial. Transpl. 27 (2012) 3191e3195.
[20] E.L. Williams, C. Acquaviva, A. Amoroso, et al., Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene, Hum. Mutat. 30 (2009) 910e917.
[21] Y. Takada, N. Kaneko, H. Esumi, P.E. Purdue, C.J. Danpure, Human peroxisomal L-alanine:glyoxylate aminotransferase:evolutionary loss of a mitochondrial targeting signal by point mutation of the initiation codon, Biochem. J. 268 (1990) 517e520. G. Rumsby / International Journal of Surgery xxx (2016) 1e6 5 Please cite this article in press as: G. Rumsby, Genetic defects underlying renal stone disease, International Journal of Surgery (2016), http:// dx.doi.org/10.1016/j.ijsu.2016.11.015
[22] P. Purdue, J. Allsop, G. Isaya, L. Rosenburg, C. Danpure, Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation, Proc. Natl. Acad. Sci. U. S. A. 88 (1991) 10900e10904.
[23] K. Hopp, A.G. Cogal, E.J. Bergstralh, et al., Phenotype-Genotype correlations and estimated carrier frequencies of primary hyperoxaluria, J. Am. Soc. Nephrol. (2015). Feb 2. pii: JASN.2014070698. [Epub ahead of print].
[24] M.J. Lumb, C.J. Danpure, Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease causing mutations, J. Biol. Chem. 275 (2000) 36415e36422.
[25] M.D. Laga, A.M.C. Pittman, A. Roncador, B. Cellini, C.L. Tucker, Allele-specific characterization of alanine:glyoxylate aminotransferase variants associated with primary hyperoxaluria, PLoS ONE 9 (2014) e94338.
[26] P.E. Purdue, Y. Takada, C.J. Danpure, Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1, J. Cell Biol. 111 (1990) 2341e2351.
[27] C.G. Monico, S. Rossetti, J.B. Olson, D.S. Milliner, Pyridoxine effect in type 1 primary hyperoxaluria is associated with the most common mutant allele, Kidney Int. 67 (2005) 1704e1709.
[28] E.C. Lorenz, J.C. Lieske, B.M. Seide, et al., Sustained pyridoxine response in primary hyperoxaluria type 1 recipients of kidney alone transplant, Am. J. Transpl. 14 (2014) 1433e1438.
[29] O. Clifford-Mobley, A. Sjogren, E. Lindner, G. Rumsby, Urine oxalate biological variation in patients with primary hyperoxaluria, Urolithiasis 44 (2016) 333e337.
[30] K. Webster, P. Ferree, R. Holmes, S. Cramer, Identification of missense, nonsense and deletion mutations in the GRHPR gene in patients with primary hyperoxaluria type II (PH2), Hum. Genet. 107 (2000) 176e185.
[31] D.P. Cregeen, E.L. Williams, S.A. Hulton, G. Rumsby, Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2, Hum. Mutat. Mutat. Brief (2003). #671 Online.
[32] E.L. Williams, E.A.L. Bagg, M. Mueller, J. Vandrovcova, T.J. Aitman, G. Rumsby, Performance evaluation of Sanger sequencing for the diagnosis of primary hyperoxaluria and comparison with targeted next generation sequencing, Mol. Genet. Genomic Med. 3 (2015) 69e78.
[33] P. Cochat, S.A. Hulton, C. Acquaviva, et al., Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment, Nephrol. Dial. Transpl. 27 (2012) 1729e1736.
[34] H.Y. Gee, I. Jun, D.A. Braun, et al., Mutations in SLC26A1 cause nephrolithiasis, Am. J. Hum. Genet. 98 (2016) 1e7.
[35] M.J. Calonge, P. Gasparini, J. Chillaron, et al., Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine, Nat. Genet. 6 (1994) 420e425.
[36] I.C. Consortium, Non-type 1 cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,þAT) of rBAT, Nat. Gen. 23 (1999) 52e57.
[37] D.S. Milliner, M.E. Murphy, Urolithiasis in pediatric patients, Mayo Clin. Proc. 68 (1993) 241e248.
[38] R. Pfeiffer, J. Loffing, G. Rossier, et al., Luminal heterodimeric amino acid transporter defective in cystinuria, Mol. Biol. Cell 10 (1999) 4135e4147.
[39] S. Nagamori, W. Wiriyasermkul, M.E. Guarch, et al., Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1, PNAS 113 (2016) 775e780.
[40] E. Pras, N. Arber, I. Aksentijevich, et al., Localization of a gene causing cystinuria to chromosome 2p, Nat. Genet. 6 (1994) 415e419.
[41] L. Bisceglia, M.J. Calonge, A. Totaro, et al., Localization, by linkage analysis, of the cystinuria type III gene to chromosome 19q13.1, Am. J. Hum. Genet. 60 (1997) 611e616.
[42] T. Eggermann, A. Venghaus, K. Zerres, Cystinuria: an inborn error of urolithiasis, Orphanet J Rare Dis. 7 (2012) 19.
[43] K. Thomas, K. Wong, J. Withington, M. Bultitude, A. Doherty, Cystinuria - a urologist's perspective, Nat. Rev. Urol. 11 (2014) 270e277.
[44] S.E. Lux, K.M. John, R.R. Kopito, H.F. Lodish, Cloning and characterization of band 3, the human erythrocyte anion-exchange protein (AE1), Proc. Natl. Acad. Sci. U. S. A. 86 (1989) 9089e9093.
[45] A.E. Schofield, P.G. Martin, D. Spillett, M.J.A. Tanner, The structure of the human red blood cell anion exchanger (EPB3, AE1, Band 3) gene, Blood 84 (1994) 2000e2012.
[46] F.C. Brossius, S.L. Alper, A.M. Garcia, H.F. Lodish, The major kidney band 3 gene product predicts an amino-terminal truncated band 3 polypeptide, J. Biol. Chem. 264 (1989) 7784e7787.
[47] L.J. Bruce, D.L. Cope, G.K. Jones, et al., Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene, J. Clin. Invest. 100 (1997) 1693e1707.
[48] N. Hooman, H. Otukesh, H. Fazilaty, I. Torktaz, R. Hosseini, B. Behnam, A novel mutation pattern of kidney anion exchanger 1 gene in patients with distal renal tubular acidosis in Iran, IJKD 9 (2015) 230e238.
[49] A.M. Toye, G. Banting, M.J.A. Tanner, Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mistargeting explains dominant renal tubular acidosis (dRTA), J. Cell Sci. 117 (2004) 1399e1410.
[50] C.Y. Chu, J. King, M. Berrini, et al., Degradation mechanism of a golgi-retained distal renal tubular acidosis mutant of the kidney anion exchanger 1 in renal cells, Am. J. Physiol. Cell Physiol. 307 (2014) C296eC307.
[51] N. Rungroj, M.A.J. Devonald, A.W. Cuthbert, et al., A novel missense mutation in AE1 causing autosomal dominant distal renal tubular acidosis retains normal transport function but is mistargeted in polarized epithelial cells, J. Biol. Chem. 279 (2004) 3833e13838.
[52] M.A.J. Devonald, A.N. Smith, J.P. Poon, G. Ihrko, F. Karet, Non-polarized targeting of AE1 causes autosomal dominant distal renal tubular acidosis, Nat. Genet. 33 (2003) 125e127.
[53] A.C. Fry, Y. Su, V. Yiu, A.W. Cuthbert, H. Trachtman, F.E. Karet Frankl, Mutation conferring apical-targeting motif on AE1 exchanger causes autosomal dominant distal RTA, J. Am. Soc. Nephrol. 23 (2012) 1238e1249.
[54] F.E. Karet, F.J. Gainza, A.Z. Gyory, et al., Mutations in the chloride-bicarbonate exhanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis, Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 6337e6342.